Polymerase chain reaction (PCR)
The repeated copying of a selected region of a DNA molecule can be performed using PCR. DNA with the target sequence is mixed with DNA polymerase, two oligonucleotide primers and nucleotides. A single starting molecule of target DNA is sufficient for the PCR to amplify large quantities of the DNA segment between the oligonucleotide primers. The primers are designed to attach to the target DNA at either side of the segment that should be amplified and are required to initiate DNA synthesis by the DNA polymerase, which incorporates the nucleotides complementary to the DNA template.
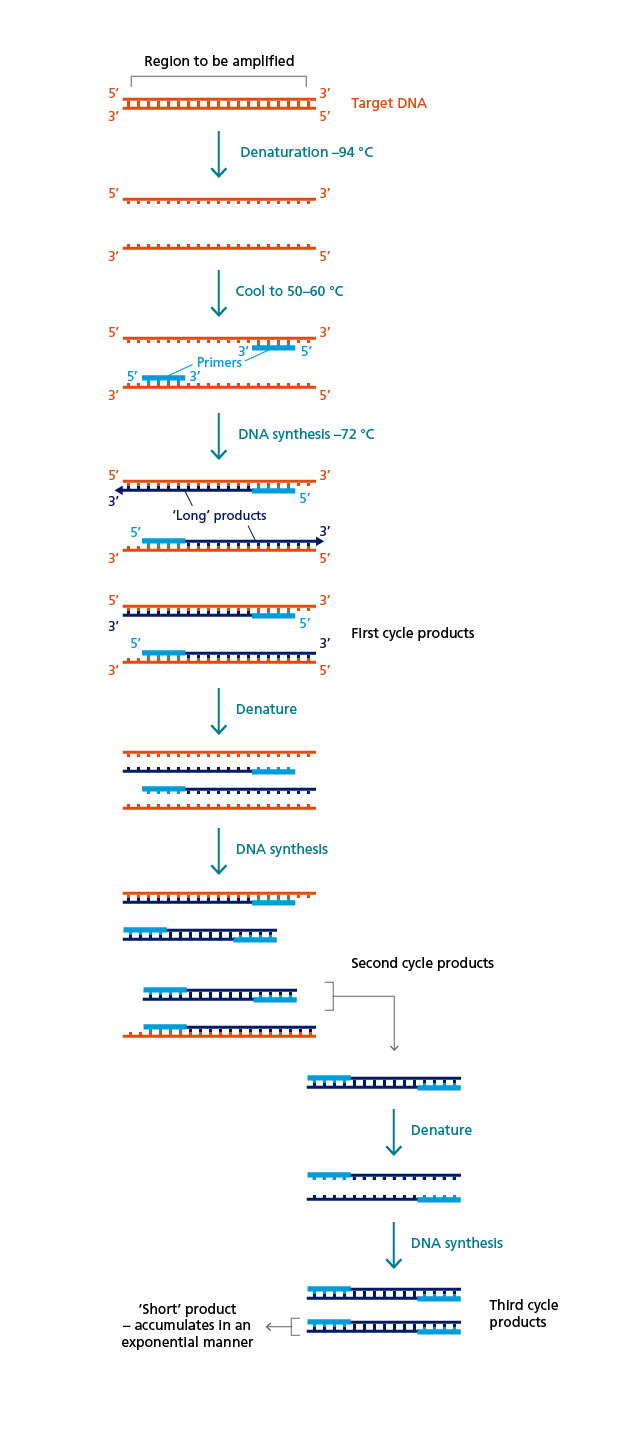
Overview of the amplification of a defined DNA sequence using PCR.
Sequencing
Although DNA sequencing is often an automated process, the method upon which it is usually based is the Sanger chain termination method. Identical single-stranded DNA molecules containing the genetic segment to be sequenced, for example amplified by PCR, are used as a template to which a short oligonucleotide primer is annealed at the same position on each molecule. The primer is the starting point for DNA polymerase to synthesise a complementary DNA strand by incorporating deoxyribonucleotide triphosphates (dNTPs). In addition to the four dNTPs, a small amount of the four dideoxynucleotides (ddNTPs), each labelled with a different fluorophore, is added to the mixture. These lead to the termination of DNA synthesis when they are incorporated into the growing chain at various positions because they lack the 3’-hydroxyl group required for subsequent polymerisation of the next nucleotide. This results in a set of chains with different lengths, all ending with one of the four fluorescently-labelled ddNTPs. The different chains are separated from one another using polyacrylamide gel electrophoresis, which can separate DNA molecules that differ in length by only a single nucleotide. The DNA sequence can be established from the sequence of the fluorescent bands on the electrophoresis gel.