Because common symptoms of von Willebrand disease (VWD) such as menorrhagia and epistaxis also occur in normal populations, the initial diagnosis of VWD can be challenging. Using questionnaires to assess bleeding symptoms and family history can help to guide assay selection, although age and other factors can affect survey results.1-5
Initial laboratory assessment of individuals with VWD can be non-specific, depending on the disease classification. Bleeding time is typically prolonged; activated partial thromboplastin time (aPTT) can be prolonged or normal depending on FVIII levels. Platelet counts are usually normal, with the exception of VWD type 2B plasma. Finally, VWD plasma often exhibits abnormal platelet function; however, mild type 1 and type 2N VWD plasma typically behaves normally in platelet function analyses.1,2
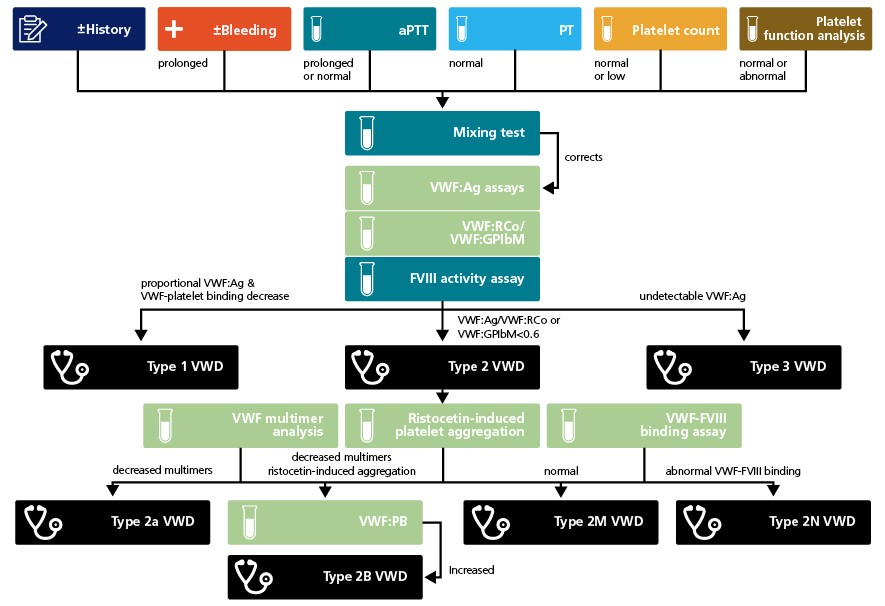
Different treatment strategies are applied depending on VWD classification. Thus, further laboratory testing beyond the initial investigation is needed to diagnose VWD and discriminate between the different types. If aPTT is prolonged, mixing tests with normal plasma can exclude the possibility of antibodies against coagulation factors, antibodies against VWF and other anticoagulants. Three additional assays are needed to diagnose and classify VWD: von Willebrand factor (VWF):antigen assay (VWF:Ag), a VWF activity assay and a FVIII activity assay. The VWF activity assay can be a VWF ristocetin cofactor assay (VWF:RCo) that uses platelets and ristocetin, an assay based on ristocetin induced binding of VWF to a recombinant wild type GPIb fragment (VWF:GPIbR) or an assay based on the spontaneous binding of VWF to a gain-of-function mutant GPIb fragment (VWF:GPIbM).6 Proportionally low VWF:Ag (VWF quantity) and VWF-platelet binding function can be indicative of type 1 VWD; however results should be interpreted with caution and ABO blood group taken into consideration because this can affect VWF levels.7 Undetectable VWF:Ag confirms a type 3 VWD diagnosis. Normal VWF:Ag and VWF:RCo or VWF:GPIbM with low FVIII activity suggest VWD type 2N or haemophilia A. If type 2 VWD is suspected, ristocetin-induced platelet aggregation, VWF multimer analysis and a VWF-FVIII binding assay can support a differential diagnosis.1,2,8
Initial laboratory screening
The initial laboratory screening for VWD often includes bleeding time, activated partial thromboplastin time (aPTT), prothrombin time (PT), platelet count, morphology and platelet function analysis. As VWD is classified into six different types according to concentration and VWF function, results of individual tests may differ depending on the disease classification.
A normal result for aPTT does not exclude VWD or mild factor (F) VIII deficiency. An isolated prolonged aPTT may indicate lower FVIII levels, secondary to a quantitative or qualitative VWD deficiency in some forms of VWD. However, coagulation factor deficiencies such as FVIII, FIX, XI or XII also present with a prolonged aPTT.
Furthermore, patients with type 2B VWD may present with a low platelet count and abnormal platelet function assessed using a platelet function analyser (PFA-100®). Abnormal PFA-100® results have also been observed in severe type 1, types 2A, 2B, 2M and type 3 VWD.1,2,8
Mixing tests
In the presence of a prolonged aPTT, an uncorrected mixing test can identify possible allo- or autoantibodies directed against coagulation factors. However, an uncorrected aPTT mixing test may also be indicative of lupus anticoagulant or acquired von Willebrand syndrome. A Betheda assay can confirm potential antibodies directed against coagulation factors and dilute Russell viper venom time (RVVT) can confirm a lupus anticoagulant.9 Generally, a VWD, mixing test should correct the aPTT immediately and after two hours.1
VWF:Ag assay, VWF activity and FVIII activity assay
VWF antigen (VWF:Ag), VWF activity, and a FVIII activity assay performed in parallel can confirm a diagnosis of VWD and help to distinguish between VWD type classifications. The VWF activity assay can be a VWF:RCo that uses platelets and ristocetin, an assay based on ristocetin induced binding of VWF to a recombinant wild type GPIb fragment (VWF:GPIbR) or an assay based on the spontaneous binding of VWF to a gain-of-function mutant GPIb fragment (VWF:GPIbM). Another option to determine VWF activity is the collagen binding assay (VWF:CB).6 Some experts recommend assessing VWF activity by its ability to bind GPIb and collagen.10 A low VWF:Ag (VWF quantity) ratio with a proportional decrease in VWF:RCo or VWF:GPIbM is considered confirmatory for type 1 VWD, with VWF:Ag <30 IU/dl considered definitive. Undetectable VWF:Ag confirms type 3 VWD. A decrease in VWF:Ag and VWF:RCo or VWF:GPIbM below normal with a ratio <0.6 or 0.7 suggest type 2A, 2B or type 2N VWD.1,2,8
Normal VWF:Ag and VWF-platelet binding function together with low FVIII activity can be indicative of haemophilia A or VWD type 2N. Abnormal VWF-FVIII binding can confirm VWD type 2N.2
VWF multimer analysis
VWF multimer analysis can distinguish between type 2M and 2A or 2B VWD. Multimers in type 2M plasma are largely normal, whereas type 2A plasma is typically missing large and intermediate multimers, and 2B plasma is usually only missing large multimers.1,2
Ristocetin-induced platelet aggregation (RIPA)
Platelet aggregation in the RIPA assay is indicative of type 2B VWD. However, mutations in the platelet VWF receptor (platelet-type VWD or pseudo-VWD) also cause platelet aggregation in the RIPA assay. An increased VWF-platelet binding assay (VWF:PB) can distinguish type 2 VWD from platelet-type VWD and pseudo-VWD.1,2
Genetic analysis
Specialist laboratories can perform a genetic analysis of the VWF gene. Analysis using sequencing may be helpful for the diagnosis of VWD types 2B, 2M and 2N, as mutations tend to be localised in specific regions of the VWF gene. Mutations that cause VWD type 3 tend to be unique to the family. Prenatal genetic analysis of VWD type 3 may be helpful for preparations during and after childbirth. However, as up to 35% of people with VWD type 1 have no characterised mutation, genetic analysis currently holds little value for diagnosis of these patients.8,11