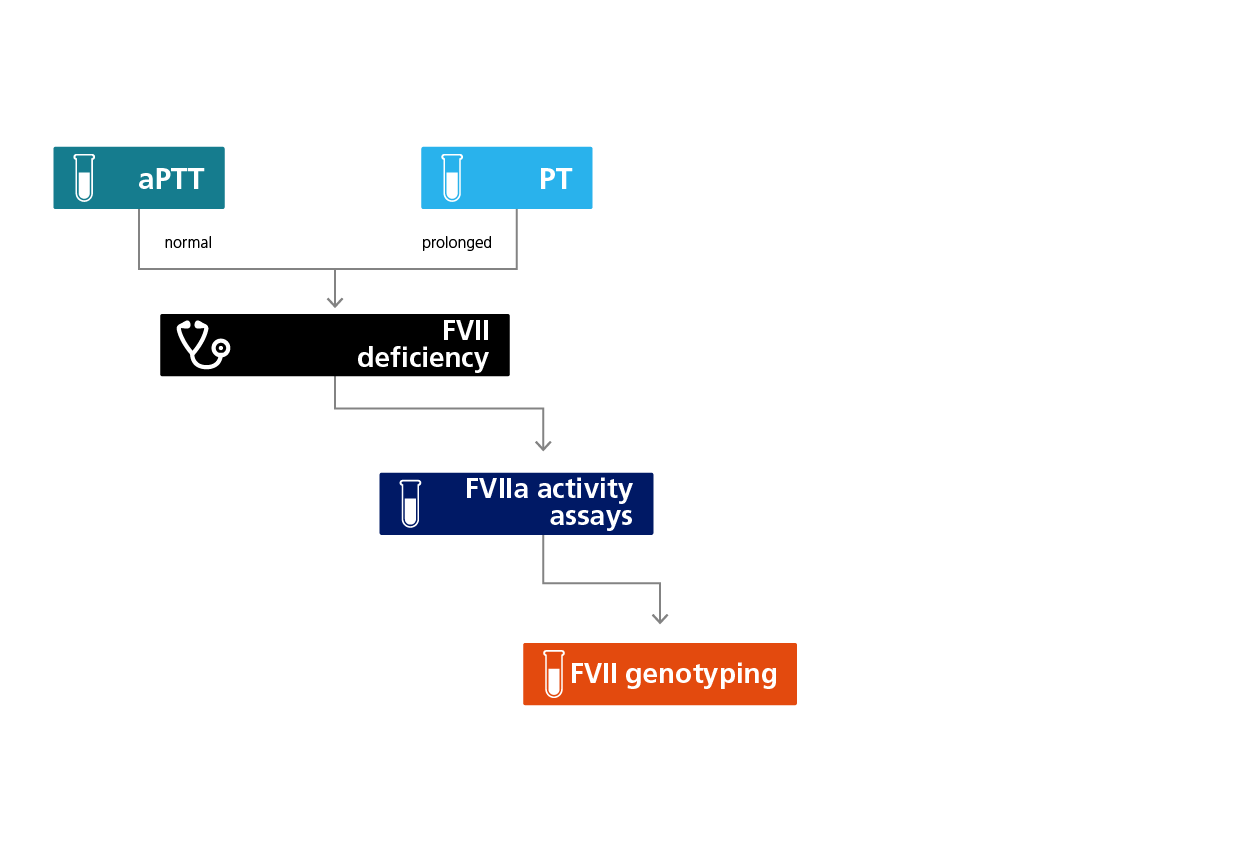
Initial laboratory screening
Results from PT and aPTT screening tests can be interpreted in
combination to identify and exclude certain coagulation disorders. A
normal aPTT excludes moderate or severe deficiencies in the extrinsic
or common coagulation pathways (i.e., FV,
FVIII, FIX, FX, FXI or FXII). An isolated, prolonged PT immediately
suggests a FVII deficiency. Prolongation of the international
normalised ratio (INR)
may depend on the level of FVII in the plasma. Aside from genetic
lesions, other factors that may influence the level of FVII activity
include liver disease, a vitamin K deficiency or treatment with
vitamin K antagonists.1,2 For some molecular defects of
FVII the abnormality is not detected with all reagents so that the PT
may be normal with one reagent but abnormal with another. In this case
the results obtained with reagents containing human tissue factor (TF)
are more likely to correlate with the clinical picture.
Factor activity assays
Activated FVII (FVIIa) is present in plasma at a concentration of
5–15 ng/mL in normal individuals, representing only about 1–3% of the
total pool of FVII, and with a half-life of 2–3 hours. FVII activity
assays serve to confirm a lack of FVII/FVIIa activity. A clot-based
assay uses a soluble, recombinant truncated TF (TFF) that is unable to
activate FVII zymogen and therefore measures FVIIa activity in a given
sample. The same assay can also be used to measure thrombin generation
or FXa activity using fluorogenic substrates. A separate
fluorogenic assay that also uses a FXa-based fluorogenic substrate
activates FVII to FVIIa and then measures the total activity.
Together, therefore, the relative proportions of activated and zymogen
FVII can be elucidated.2,3
The choice of assay reagents to measure clot assay-based FVII
activity is critical, because the origin of the thromboplastin used in
the assay may influence the results. In addition, residual amounts of
FVII activity within factor-‘deficient’ plasma may reduce the
sensitivity of assays in the lower activity range. Assay calibration
using a plasma standard and the introduction of recombinant TF and
synthetic phospholipid preparations have reduced the inter-laboratory
variability associated with both of these components.1-3
Molecular analysis (genotyping)
A molecular genetic analysis of the FVII gene (F7) represents
the gold standard to confirm a diagnosis of FVII CD. A full F7
sequence following conventional PCR
amplification identifies the genetic lesion in most cases. The
analysis is also useful in the presence of a discordant bleeding
phenotype and FVII activity or an unclear inheritance pattern. A
pre-natal diagnosis using umbilical cord blood in the presence of a
family history or bleeding is also possible.1
Immunological FVII antigen assays
Zymogen-activation-based deficiencies can be confirmed or excluded
using available FVII immunological enzyme-linked immunosorbent assays
(ELISA)
to distinguish between FVII zymogen and FVIIa.