Because individuals with hereditary FXI deficiency (FXI CD) frequently show only mild or no bleeding symptoms unless challenged by some types of surgery or other trauma, a diagnosis may be delayed and/or uncovered incidentally during pre-surgical laboratory screening. A more detailed medical anamnesis may reveal a history of epistaxis or menorrhagia that are consistent with a mild bleeding disorder.
There is little association between FXI plasma activity and bleeding risk or clinical severity, however other factors, such as the balance between fibrinolysis and thrombin-activatable fibrinolysis inhibitor (TAFI) activity may play a contributing role. Routine laboratory assessments are therefore not able to predict the risk of bleeding, however invasive procedures at sites with high levels of fibrinolysis such as the genitourinary tract and oral cavities should be approached with caution.1-3
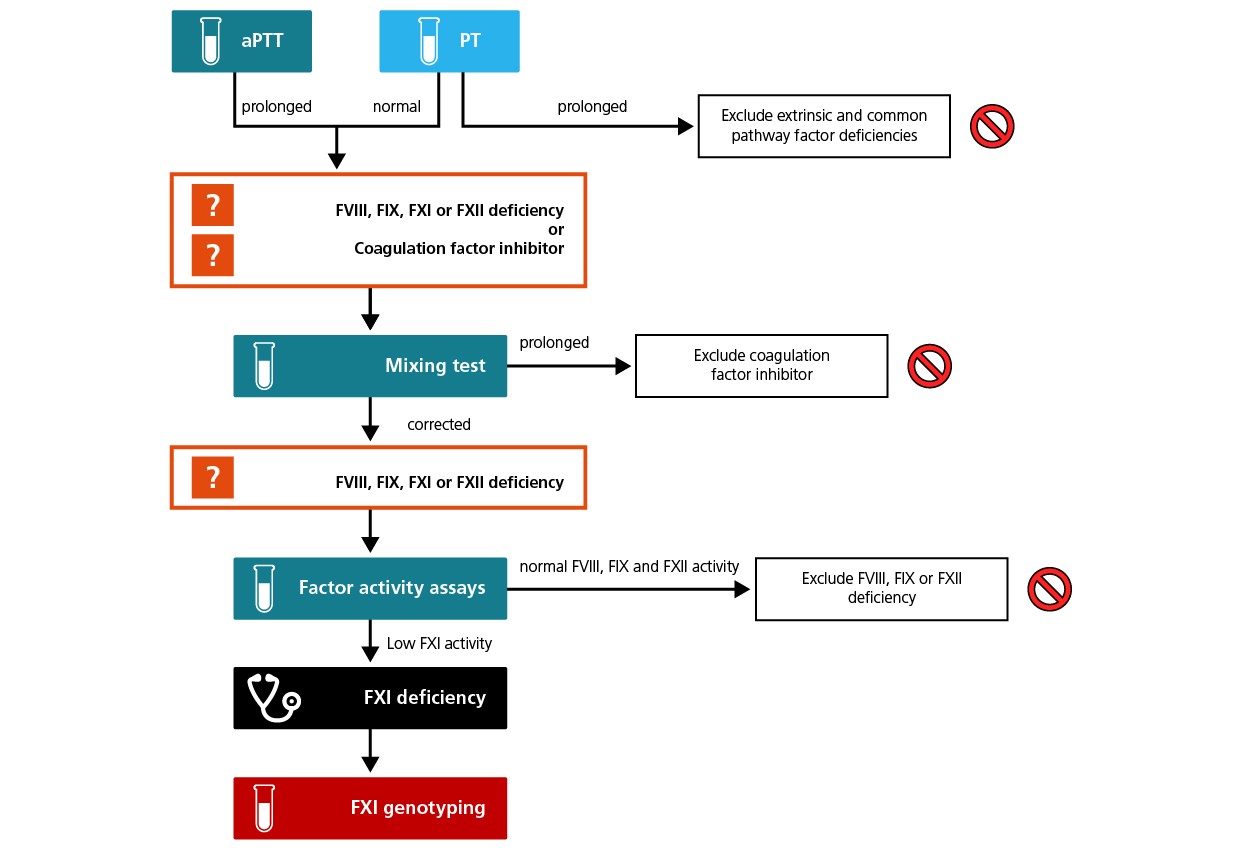
A prolonged activated partial thromboplastin time (aPTT) along with a normal prothrombin time (PT) and bleeding symptoms are suggestive of a FXI deficiency once FVIII deficiency, FIX deficiency, and von Willebrand disease have been excluded. The presence of a lupus anticoagulant or a FXII deficiency to explain the isolated prolonged aPTT should also be ruled out.4,5
Both thrombin generation assays that block the contact activation of FXI by FXIIa and thromboelastometry have been used on an experimental basis to assess thrombin-induced FXI activity and a possible correlation with bleeding risk. The inactivation of tissue factor pathway inhibitor (TFPI) by FXIa suggests that FXI plays a role in maintaining haemostasis by tempering the activity of TFPI. Accordingly, TFPI may represent a marker for bleeding risk in patients with severe FXI CD.1,2,4
Women with either homozygous of heterozygous FXI CD frequently experience menorrhagia, however this is seldom the presenting symptom that leads to a diagnosis. FXI levels do not change during pregnancy and FXI CD is not associated with an increased rate of prepartum complications. However, women with a family history of FXI CD and/or from populations with known FXI CD founder effects should be routinely screened using an aPTT assay and haemostatic treatment considered during epidural anaesthesia and delivery.4,6
Although circumcision is not typically associated with excessive bleeding in males with FXI CD, a screen for cord blood FXI plasma activity in newborns at risk may be prudent, especially if the procedure is performed outside a medical facility or by non-medical personnel. The level of plasma FXI present at birth is lower than in older children and adults, potentially due to reduced hepatic function, therefore a delay of at least six months and re-testing of FXI activity may provide a more accurate measure of disease severity.4,6,7
Patients with severe FXI CD and a very low level of plasma FXI who have been exposed to blood products containing FXI, typically fresh frozen plasma, FXI concentrate or immunoglobulins, including anti-Rhesus factor immunisation, are at risk of developing alloantibodies to FXI. While these inhibitor patients may not bleed spontaneously, they may experience prolonged bleeding during or following surgery due to neutralisation of any endogenous or exogenous FXI. Screening for potential FXI alloantibodies in patients with null mutations previously exposed to FXI is therefore prudent.4,7
The >200 heterogeneous genetic mutations identified to date are distributed throughout the FXI gene on chromosome 4, including the coding sequence, splice junctions and promoter region. Once common mutations and large deletions have been excluded using PCR-based screening methodology, a full mutational sequence analysis for patients from populations without a known founder effect may be necessary. Identification of the genetic anomaly is particularly important in patients with very low or no detectable FXI plasma activity in order to estimate the risk of alloantibody development.4
Initial laboratory screening
Routine laboratory testing usually includes an activated partial thromboplastin time (aPTT), and individuals with hereditary FXI CD consistently show a prolonged aPTT. The aPTT alone, however, is not diagnostic, as deficiencies in other intrinsic coagulation factors such as FVIII, FIX, FXII and von Willebrand factor or a lupus anticoagulant may also prolong the aPTT result.
Different aPTT reagents are associated with different levels of sensitivity, therefore, local reference ranges should be established with plasma from non-affected individuals. Depending on the level of sensitivity of the reagent used, a normal aPTT result may not exclude a FXI deficiency.1,4,7
aPTT screening tests assess the initial rate of clot formation, however because FXI appears to play a role in the consolidation and maintenance of clot formation after the initial thrombin burst, this assay may not adequately assess the effect of FXI activity. In addition, contact activation of FXII in the aPTT reaction may misrepresent physiological conditions and the importance of FXI activation by FXIIa.1
Mixing tests
Suspicion of a FXI deficiency can be solidified using a mixing test to show that patient plasma fails to correct the aPTT of FXI-deficient plasma.
An aPTT-based mixing test can also be used to identify the presence of a potential alloantibody, which can be confirmed using the Bethesda assay.4,6
Factor activity assay
The diagnosis is confirmed using a FXI activity assay to quantify the deficiency. This is usually done using a one-stage assays based on aPTT.4
Immunological FXI antigen assay
A FXI enzyme-linked immunoabsorbant assay (ELISA), if available, can be used to distinguish between quantitative and qualitative FXI deficiencies.4,6
Molecular analysis (genotyping)
In the presence of a family history of FXI CD or an ethnic background with a known founder effect, the molecular anomaly associated with the FXI deficiency can be identified or confirmed using polymerase chain reaction-based assays to amplify the region near the suspected mutation. In the absence of a family history of FXI CD or the potential presence of a known founder mutation to focus the assessment, genetic analysis may require a full molecular sequence of the FXI gene. Large heterozygous deletions and new mutations, especially those in the non-coding regions of the gene that affect transcription or splicing, may be particularly challenging to detect. Novel mutations should be tested in silico and in vitro to confirm the functional pathogenic effect.4,5
1. Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol 2016;9:629-37.
2. Puy C, Rigg RA, McCarty OJ. The hemostatic role of factor XI. Thromb Res 2016;141 Suppl 2:S8-S11.
3. Peyvandi F, Palla R, Menegatti M, et al. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost 2012;10:615-21.
4. Duga S, Salomon O. Congenital factor XI deficiency: an update. Semin Thromb Hemost 2013;39:621-31.
5. Duga S, Salomon O. Factor XI Deficiency. Semin Thromb Hemost 2009;35:416-25.
6. Gomez K, Bolton-Maggs P. Factor XI deficiency. Haemophilia 2008;14:1183-9.
7. Bolton-Maggs PH. Factor XI deficiency--resolving the enigma? Hematology Am Soc Hematol Educ Program 2009:97-105.