
Hemofilia B
La hemofilia B (HB; 2017 ICD-10-CM: D67) es un trastorno de la coagulación hereditario, cuya causa es la falta del factor (F) de coagulación IX. El gen F9 codifica para el FIX, que media la conversión enzimática de FX a la forma activada FXa. Como consecuencia de esta deficiencia congénita, los pacientes con HB presentan un proceso de coagulación defectuoso y una mayor tendencia a sufrir hemorragias de manera espontánea o en respuesta a una lesión traumática. El tratamiento estándar para prevenir y controlar las hemorragias consiste en el reemplazo del factor de coagulación faltante.
La HB representa aproximadamente el 10% de todos los trastornos de la coagulación y tiene una prevalencia de alrededor de 3 de cada 100.000 hombres en países de altos ingresos económicos.1,2
La prevalencia de HB reportada difiere según el estado económico de un país.3 A nivel mundial, alrededor del 70% de los pacientes con hemofilia están mal diagnosticados y no reciben tratamiento; la mayoría de estos pacientes viven en países en vías de desarrollo.3
Las manifestaciones clínicas de la HB son parecidas a las de la hemofilia A e incluyen hemorragias prolongadas y repetidas. A menos que se reciba el tratamiento adecuado, las hemorragias en articulaciones y músculos producen artropatías progresivas dolorosas y atrofia muscular,4 y pueden derivar en una discapacidad grave con secuelas profesionales y psicológicas. Si bien los sitios más frecuentes donde se producen las hemorragias espontáneas son las articulaciones y los músculos,5 las hemorragias en el sistema nervioso central son la principal causa de muerte en los pacientes con hemofilia, que no están infectados con el virus de la inmunodeficiencia humana (VIH), lo que representa aproximadamente un tercio de todas las muertes.6
La HB se clasifica en leve, moderada o grave según la actividad del FIX residual.5 Los pacientes con HB leve presentan hemorragias graves durante cirugías o traumatismos de importancia, pero rara vez sufren hemorragias espontáneas. Los pacientes con HB moderada experimentan hemorragias ocasionales y prolongadas con cirugías o traumatismos menores. La HB grave se caracteriza por hemorragias espontáneas en las articulaciones o músculos, principalmente en ausencia de desafíos hemostáticos identificables. La actividad del FIX residual se suele expresar como un porcentaje de la coagulación "normal", que varía entre individuos y puede oscilar entre 50 y 150%.7
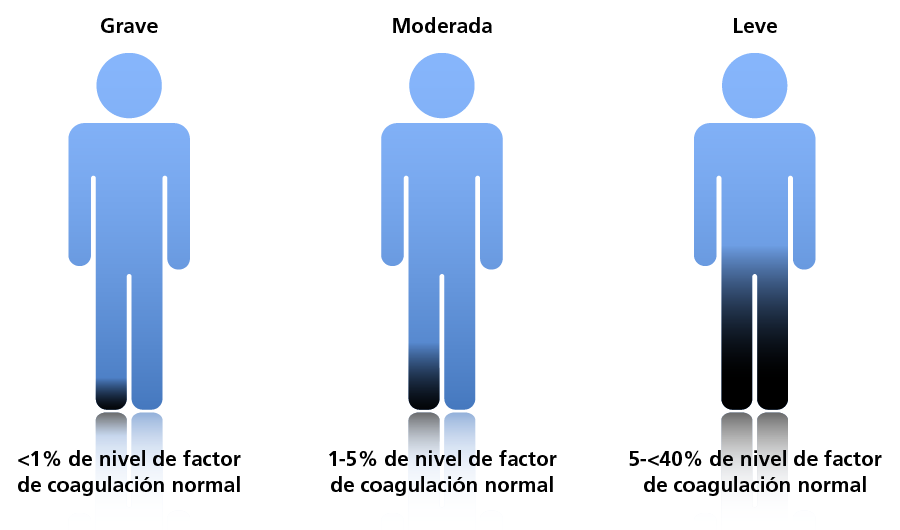
Clasificación de la hemofilia B según los niveles de factor de coagulación.5
La HB se caracteriza por distintos defectos moleculares subyacentes que afectan al gen F9. La hemofilia congénita se transmite de manera recesiva ligada al cromosoma X. No obstante, en un tercio de los casos aproximadamente, no existen antecedentes familiares y la hemofilia se produce por una mutación espontánea.8 Este equilibrio entre pérdida del gen causada por una baja idoneidad reproductiva y la mutación espontánea ha mantenido la hemofilia dentro de la población general.
Se sabe que una gran cantidad de mutaciones genéticas afectan la expresión del factor de coagulación, entre ellas mutaciones contrasentido, terminadoras (o sin sentido), defectos en el empalme de mRNA, inserciones y deleciones.8 La Lista de mutaciones del F9 del Proyecto de mutaciones de la hemofilia B (CHAMP) de los CDC ofrece un panorama general de más de 1000 mutaciones únicas identificadas hasta la fecha para la codificación del gen F9 para el FIX.9
- Stonebraker JS, Bolton-Maggs PH, Michael Soucie J, Walker I, Brooker M. A study of variations in the reported haemophilia B prevalence around the world. Haemophilia 2012;18:e91-4.
- World Federaton of Hemophilia (WFH): Report on the Annual Global Survey 2015; 2016.
- O'Mahony B, Black C. Expanding hemophilia care in developing countries. Semin Thromb Hemost 2005;31:561-8.
- Luck JV, Jr., Silva M, Rodriguez-Merchan EC, Ghalambor N, Zahiri CA, Finn RS. Hemophilic arthropathy. J Am Acad Orthop Surg 2004;12:234-45.
- World Federation of Hemophilia (WFH): Guidelines for the Management of Hemophilia; 2012.
- Darby SC, Kan SW, Spooner RJ, et al. Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood 2007;110:815-25.
- Moser KA, Adcock Funk DM. Chromogenic factor VIII activity assay. Am J Hematol 2014;89:781-4.
- Goodeve AC. Hemophilia B: molecular pathogenesis and mutation analysis. J Thromb Haemost 2015;13:1184-95.
- Li T, Miller CH, Payne AB, Craig Hooper W. The CDC Hemophilia B mutation project mutation list: a new online resource. Mol Genet Genomic Med 2013;1:238-45.
HQ19H00001, date of preparation: February 2019