Factor XI congenital deficiency (FXI CD; 2019 ICD-10-CM: D68.2) is an autosomal recessive inherited bleeding disorder caused by reduced levels of coagulation factor FXI. FXI CD is inherited in an autosomal recessive manner, has a prevalence of approximately 1:1,000,000 individuals irrespective of sex and is found in all ethnic and racial populations. Certain mutations, however, are more common among some ethnic populations due to founder effects. Communities of Ashkenazi Jewish heritage show the highest prevalence of severe FXI deficiency, with one among every 450 persons affected and as much as 5–10% of the population heterozygous carriers.1-3
Other names associated with the disorder are Rosenthal syndrome, plasma thromboplastin antecedent deficiency and haemophilia C.
Plasma FXI circulates as a homodimer with high molecular weight kininogen (HMWK) and exhibits a half-life of approximately 50 hours. FXIIa activates FXI by cleavage (FXIa) exposing the substrate binding site on the FXIa homodimer catalytic domain. 2,4
According to the traditional coagulation cascade model, FXI serves as the link between the contact activation pathway and FIX activation. This initial understanding is attributable to the aPTT assays used to study coagulation in vitro, in which FXII is activated by contact with surfaces or assay components and in turn activates FXI. However, it is now apparent that the primary physiological activator of FXI is thrombin (FIIa) on the surface of platelets, following the initial tissue factor-FVIIa-induced burst upon vascular tissue injury. Thrombin-induced activation of FXI is enhanced by the presence of anionic polymers of phosphate groups (polyphosphates), which represent a likely co-factor in vivo. FXIa is also able to autoactivate. While thrombin-induced FXI activation appears to play a role in haemostasis, FXIIa-mediated FXI activation is not required for haemostasis in vivo.1,3-6
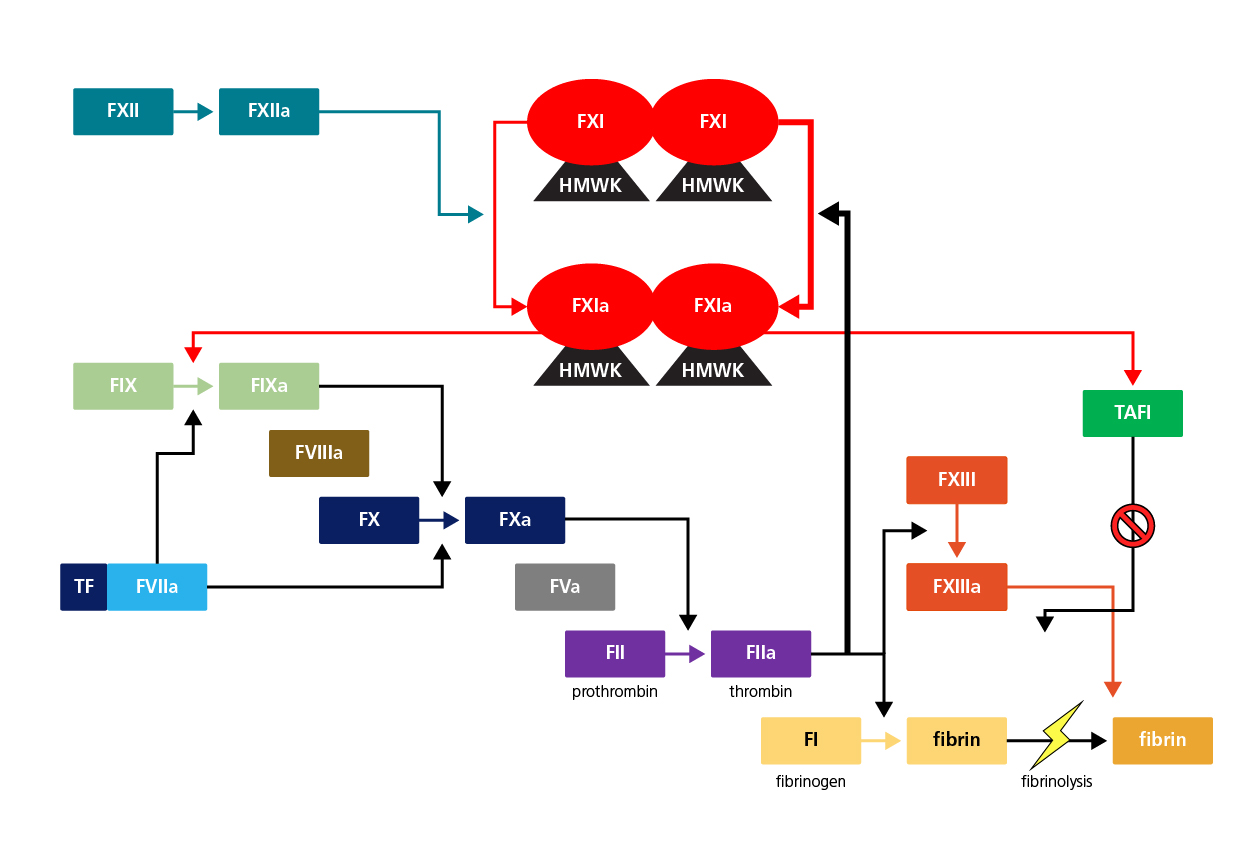
The role of FXI in haemostasis and fibrinolysis.
TF, tissue factor
HMWK, high
molecular weight kininogen
TAFI, thrombin-activatable
fibrinolysis inhibitor
FXI is not an essential component of the thrombin generation cascade, therefore its primary function may lie elsewhere, including the physiological response to injury or injection.1,4,7 FXI may play a more important role in thrombosis than haemostasis. Patients with severe FXI CD appear to be protected from ischaemic stroke, but not myocardial infarction, and exhibit a lower incidence of venous thromboembolism. Conversely, individuals with a high level of FXI activity, endogenous or exogenous, may be at higher thrombotic risk.2,5
Over 200 mutations in the FXI gene (chromosome 4) have been identified, however two mutations account for >90% of the dysfunctional alleles among people of Ashkenazi Jewish heritage. One of these mutations (Glu117stop; type II) results in a null phenotype while the other (Phe283Leu; type III) interferes with dimerisation and reduces protein secretion. The type II mutation is also found in other Jewish and Arab populations with origins in North Africa and the Middle East. Specific other mutations are associated with a founder effect among French Basques, and populations elsewhere in France, Italy, the UK and Korea. 1-3
Most FXI mutations interfere with protein synthesis, dimerisation, secretion or function, resulting in low plasma activity. Heterozygous individuals may express intermediate levels of activity. Although the disorder is inherited in an autosomal recessive manner, some mutations in heterozygous individuals may result in heterodimers comprising a wild type and a mutant protein that are not secreted, resulting in a dominant negative effect that contributes to the observed phenotypic variability. Autosomal dominant mutations are generally more common among populations of non-Jewish ancestry.1,2,8
Patients who lack FXI activity exhibit variable but relatively mild bleeding symptoms, often only during surgical procedures or other trauma. As a result, patients may be diagnosed incidentally, late in life or not at all. Spontaneous, soft tissue and joint bleeding are not typical of FXI CD, however epistaxis and menorrhagia in women are common, as is occasional peripartum bleeding. Surgical procedures that impact tissues with relatively high local fibrinolytic activity such as the oral cavity, tonsils, nasal and urogenital tracts are most likely to be associated with bleeding, while surgery or traumatic injury to other body parts may not bleed. Bleeding is most likely to manifest in tissues with a high level of fibrinolytic activity because FXI indirectly enhances the activity of thrombin-activatable fibrinolysis inhibitor (TAFI), which reduces fibrinolysis and results in more stable clot formation.2,3
Patients with <15–20% of normal FXI activity are classified as having severe FXI deficiency and those with between 20–65% of normal activity a mild or partial deficiency. However, bleeding tendency correlates poorly with factor activity, and even patients with severe deficiency may not bleed without provocation.
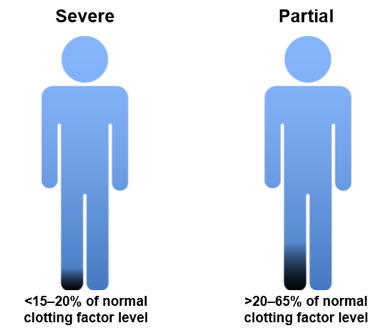
Classification of FXI CD according to clotting factor levels.
The individual bleeding phenotype may be influenced by the activity of other components of the coagulation system, including the tissue factor-FVIIa response and fibrinolysis, which may either compensate for or expose reduced FXI activity. The haemostatic role of FXI is therefore ancillary and may not even be required in some individuals. Different genetic mutations may also contribute to the variability in clinical phenotype, such that even heterozygous individuals exhibit a bleeding tendency, particularly in the presence of a dominant negative effect. Because FXI is produced by hepatocytes, individuals with reduced liver function may have lower levels of FXI expression.1-3,9
