The clinical manifestations of HB are similar to those of haemophilia A, and include prolonged and repeated bleeding episodes. Unless properly treated, haemorrhage into joints and muscles causes painful, progressive arthropathy and muscular atrophy,4 and can result in severe disability with vocational and psychological sequelae. While joints and muscles are the most common spontaneous bleeding sites,5 central nervous system bleeds are the leading causes of death in patients with haemophilia, who are not infected with human immunodeficiency virus (HIV), accounting for approximately one-third of all deaths.6
HB is classified as 'mild', 'moderate' or 'severe' according to the residual activity of FIX.5 Patients with mild HB have severe bleeding during major trauma or surgery, but rarely have spontaneous bleeding. Patients with moderate HB experience occasional bleeding and prolonged bleeding with minor trauma or surgery. Severe HB is characterised by spontaneous bleeding into joints or muscles, predominantly in the absence of identifiable haemostatic challenge. Residual FIX activity is generally expressed as a percentage of ‘normal’, which is variable between individuals and may range from 50–150%.7
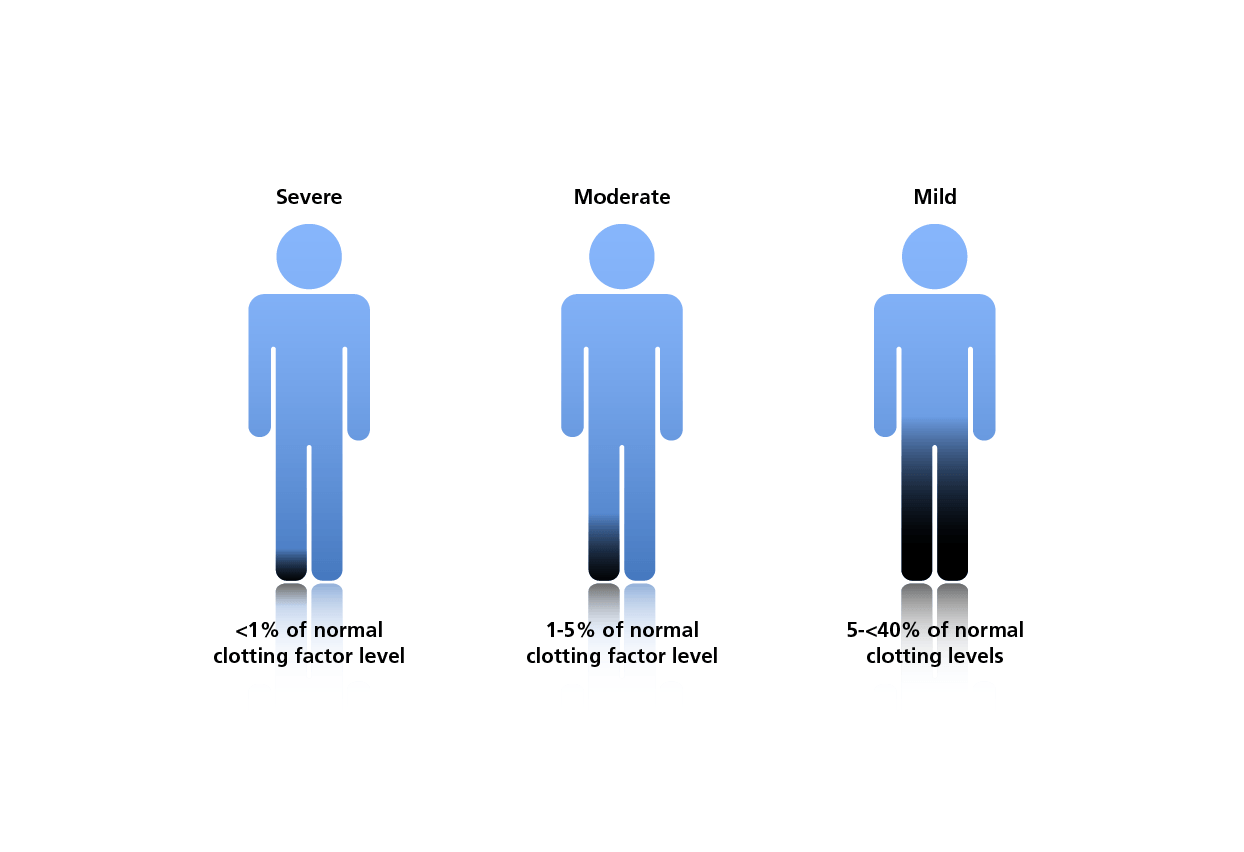
Classification of haemophilia B according to clotting factor levels5
HB is characterised by different underlying molecular defects affecting the F9 gene. Congenital haemophilia is inherited in an X-linked recessive manner. In approximately one-third of patients, however, there is no family history and haemophilia results from spontaneous mutation.5 This balance between gene loss caused by low reproductive fitness and spontaneous mutation has sustained haemophilia within the general population.
A large number of gene mutations are known to affect clotting factor expression, including missense or nonsense mutations, defects in mRNA splicing, insertions and deletions.8 The CDC Hemophilia B Mutation Project (CHBMP) F9 Mutation List provides an overview of more than 1000 unique mutations identified to date for the F9 gene encoding FIX.9
